
来源:科诺科研
尼龙6作为全球年产量达600万吨的关键工程塑料,其传统生产路线完全依赖石化原料。随着碳中和目标的推进,利用可再生生物质资源替代石油成为研究热点。木质素作为植物细胞壁的主要组分,是自然界最丰富的芳香族聚合物,其解聚得到的酚类生物油本应是制造尼龙6前体——环己酮的理想原料。然而,这一路线面临双重内在难题:一方面,酚类化合物中芳基-甲氧基键的解离能高达409-421 kJ·mol⁻¹,远高于烷基醚中的C-O键(约350 kJ·mol⁻¹),需要高温高压氢气才能断裂;另一方面,目标产物环己酮中的C=O键在苛刻条件下极易被过度加氢生成环己醇或环己烷,导致选择性丧失。这种“脱甲氧基化”与“酮基保留”之间的本质性选择权衡,使得从木质素直接制备环己酮成为长期未解的挑战。
针对上述难题,浙江工业大学林丽利教授、何荟文博士和北京大学马丁教授、浙江大学姚思宇教授合作开发了一种无氢光热催化策略,利用RuPd/TiO₂双金属催化剂在150 ℃温和条件下,以94%的选择性和12 mol·molRu+Pd⁻¹·h⁻¹的活性,将木质素衍生的酚类生物油选择性转化为环己酮类化合物(图1b-III)。该反应通过质子耦合电子转移过程,利用光生电子驱动芳基-甲氧基键的精准断裂,同时将甲氧基和水作为原位氢源。这种原位生成的低浓度活性氢物种是维持高选择性的关键。研究团队进一步将烷基取代的环己酮转化为相应的己内酰胺单体,并与常规己内酰胺共聚,获得了丙基功能化的聚酰胺6共聚物。与常规尼龙6(透明率71.5%、断裂伸长率120%)相比,新型共聚物的透光率提升至88.9%,断裂伸长率飙升至523%,展现出突破性的透明性和弹性。相关论文以“Photothermal catalytic synthesis of cyclohexanones from lignin-derived phenolic bio-oils enabling transparent and elastic polyamides”为题,发表在Nature Communications上。

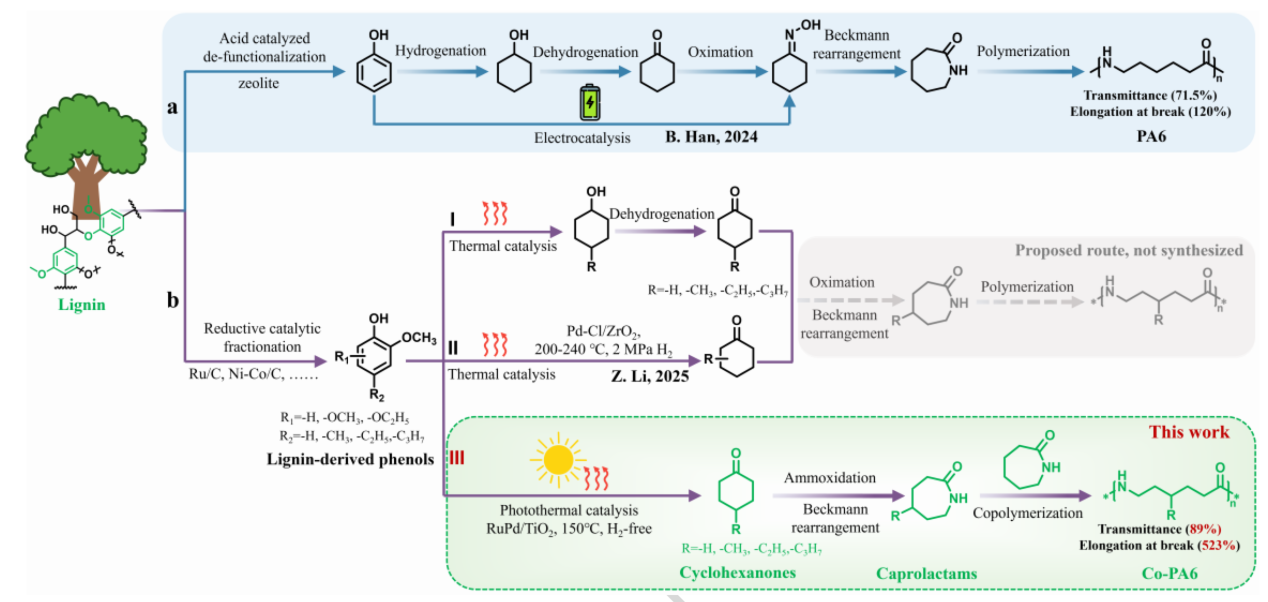
图1 | 生物基PA6的合成路线。 a. 通过酸催化木质素去官能化得到苯酚,随后转化为环己酮并进一步转化为己内酰胺的生物基PA6合成通用策略(路线a);b. 通过还原催化分馏获得的木质素衍生酚类(如取代愈创木酚)的直接增值化利用(路线b),其中路线b-I展示了传统热催化通常存在的过度加氢生成环己醇的问题,路线b-II展示了需要苛刻条件(如200-240°C,2 MPa H₂)的直接环己酮生产,路线b-III(本工作)展示了一种无氢光热催化策略,用于选择性合成生物基环己酮衍生物以及生产透明和弹性丙基官能化共聚酰胺(Co-PA6)。灰色虚线区域突出显示了烷基官能化聚酰胺合成与性能评估这一尚未充分探索的领域。
研究团队首先以木质素模型化合物愈创木酚为底物,系统评估了光热催化体系的性能。在150 ℃全光谱光照下,愈创木酚被选择性转化为环己酮作为主要产物(图2a)。RuPd/TiO₂双金属催化剂展现出远优于单金属Ru/TiO₂和Pd/TiO₂以及两者物理混合催化剂的活性,环己酮生成速率显著提高(图2b)。为验证光热协同效应的独特优势,研究者对比了光照条件下的光热催化与黑暗条件下的纯热催化。结果令人瞩目:在无外加氢气的黑暗热催化条件下,环己酮选择性极低,产物以过度加氢的环己醇为主;而在光照下,即使不外加氢气,也能以高选择性获得环己酮。更有意思的是,当在光照体系中引入0.37 MPa的氢气时,环己酮选择性反而大幅下降,这充分证明了原位生成的“低浓度”活性氢是维持高选择性的关键(图2c)。该催化剂还表现出优异的循环稳定性,多次重复使用后活性和选择性均无明显衰减(图2d)。
为揭示光热催化过程的微观机制,研究团队进行了一系列精细的机理实验。采用不同捕获剂的对照实验揭示了反应的关键步骤:加入强空穴捕获剂Na₂SO₃后反应几乎被完全抑制,而加入电子捕获剂Ag⁺也表现出显著的抑制效应,证明光生空穴和电子都是不可或缺的参与者。相反,加入·OH自由基捕获剂异丙醇对转化率影响甚微,但加入非选择性自由基捕获剂TEMPO后反应被有效终止,表明环己酮的生成是通过自由基路径完成的(图2e)。更为重要的是,利用TEMPO捕获并结合液相色谱-高分辨质谱分析,研究者成功鉴定出两个关键自由基中间体——脱氢愈创木酚自由基和脱氢环己酮自由基,这为理解反应路径提供了直接证据(图2f)。基于上述结果,研究团队提出了合理的反应机制:光照下催化剂产生电子-空穴对,肖特基结促进其高效分离;光生空穴优先氧化吸附在催化剂表面的酚类底物中的甲氧基,在水分子协助下断裂Ar-OCH₃键,同时从水和甲氧基中原位产生活性氢物种;这些表面活性氢随后对芳香环进行选择性加氢,从而在脱甲氧基化和加氢之间建立动力学平衡,有效防止酮基的过度加氢(图2g)。
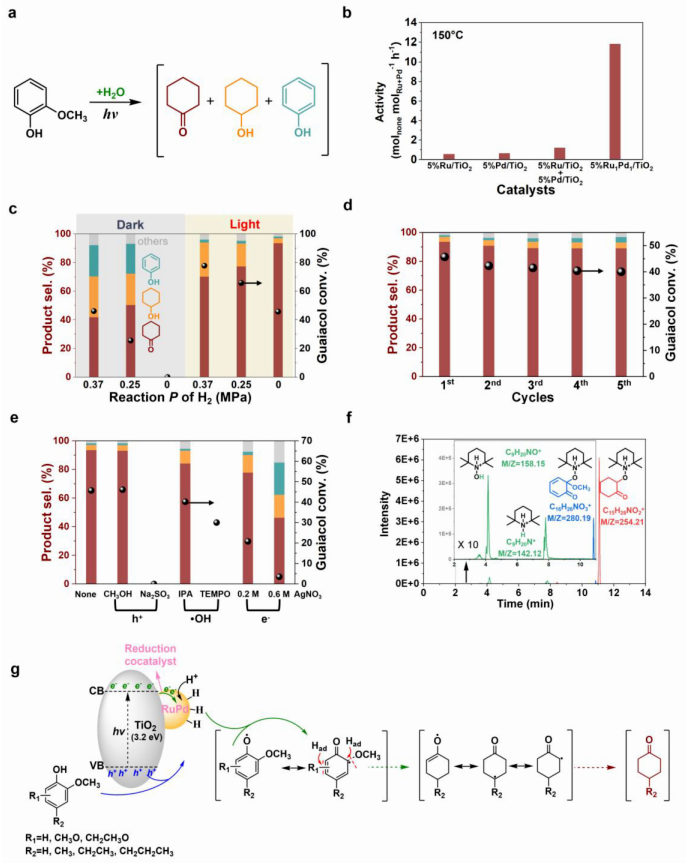
图2 | 愈创木酚光热催化转化的催化性能评估与机理研究。 a. 愈创木酚光热选择性转化的主要产物;b. 在150°C全光谱光照射下,5% Ru/TiO₂、5% Pd/TiO₂、5% Ru/TiO₂+5% Pd/TiO₂物理混合催化剂以及双金属5% RuPd/TiO₂催化剂上的光热反应活性(环己酮生成速率);c. 在150°C全光谱光照射(320-780 nm)下,不同氢气压力条件下5% Ru₁Pd₁/TiO₂催化剂上愈创木酚转化的光热催化性能(右),与黑暗条件下热催化性能的对比(左);d. 5% Ru₁Pd₁/TiO₂催化剂在150°C全光谱光照射下用于愈创木酚光热催化转化为环己酮的循环稳定性;e. 在5% Ru₁Pd₁/TiO₂光催化剂上进行愈创木酚转化时,存在电子、空穴和自由基捕获剂的对照实验;f. 在TEMPO(15 mmol/L)存在下,愈创木酚光催化反应后液相产物中产物的典型液相色谱和质谱图;g. 所提出反应机理的示意图。反应条件:底物(4 mmol)、催化剂(0.10 g)、H₂O(25 mL)、150°C、1000 rpm搅拌、3小时,辐射强度为0.35 W/cm²(氙灯光源)。
为验证该光热催化策略的普适性,研究团队选取了八种代表性木质素衍生的酚类化合物进行测试。令人振奋的是,所有底物在相同反应条件下均表现出超过80%的环己酮及其烷基化衍生物选择性,转化率介于40.6%至66.2%之间。更重要的是,烷基侧链得以完整保留,而非在反应中被脱除——这恰恰体现了该策略的精妙之处,即保留木质素天然碳骨架的烷基结构,为后续赋予聚合物特殊功能奠定基础(图3a)。研究团队进一步挑战了更为复杂的混合底物体系,将上述八种化合物等摩尔混合后进行一锅反应,依然获得了86.7%的高选择性和42.0%的转化率,充分证明了该催化体系处理复杂多组分底物的鲁棒性(图3b)。最具说服力的是对真实木质素生物油的验证。研究者采用还原催化分馏法从桦木木质纤维素中提取真实生物油,其主要成分为4-丙基愈创木基和4-丙基紫丁香基单体。尽管原料中包含复杂的木质纤维素衍生物杂质,催化剂仍高效选择性地将其中主要组分转化为4-丙基环己酮,累积产量达44.1 mg。气相色谱-质谱谱图清晰地展示了木质素生物油原料的组成以及最终光热反应混合物的产物分布(图3c)。
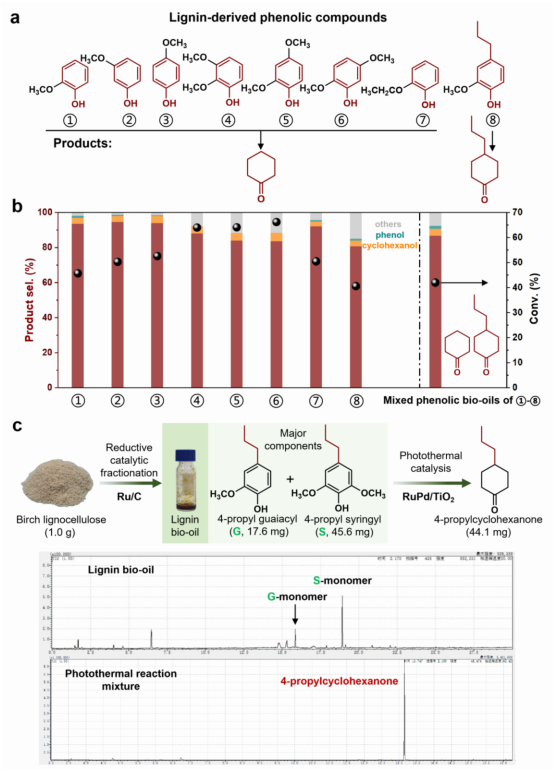
图3 | 无氢光热催化策略的普适性与实际应用性。 a. 底物范围考察;化合物1-7的主要产物为环己酮,而化合物8的主要产物为烷基环己酮;b. 单个底物(1-8)和混合酚类生物油的催化性能(选择性和转化率)。混合酚类生物油中八种化合物的摩尔比为1:1:1:1:1:1:1:1。反应条件:底物(4 mmol)、催化剂(0.10 g)、H₂O(25 mL)、150°C、1000 rpm搅拌、3小时,辐射强度为0.35 W/cm²(氙灯光源);c. 来自桦树木质纤维素的真实木质素生物油的实际增值化利用。示意图展示了通过还原催化分馏过程生产木质素生物油(富含4-丙基愈创木基(G)和4-丙基紫丁香基(S)单体),随后在5% Ru₁Pd₁/TiO₂光催化剂上进行顺序批次光热催化,累计得到4-丙基环己酮(44.1 mg)。GC-MS色谱图(底部)确认了木质素生物油原料和最终光热反应混合物的组成。详细反应条件见补充信息(补充表4)。
受真实木质素生物油中富含丙基取代酚类化合物并能够选择性转化为4-丙基环己酮的启发,研究团队进一步探索了烷基功能化聚酰胺材料的合成与性能。他们以商业4-丙基环己酮为原料,通过肟化和贝克曼重排两步反应获得高纯度4-丙基己内酰胺单体,并将其与常规己内酰胺按不同比例共聚,获得了一系列丙基功能化共聚酰胺(图4a)。差示扫描量热分析表明,随着4-丙基己内酰胺插入比例增加,共聚物的熔点和结晶度显著下降——15%插入比例时结晶度从常规尼龙6的36.7%降至19.6%,当功能单体比例超过30%时共聚物完全变为无定形态(图4b)。二维广角X射线衍射进一步证实,丙基支链的存在有效破坏了氢键网络,随着丙基含量增加,衍射环逐渐变宽,表明链间有序排列被破坏(图4c)。
丙基功能化单体的引入为共聚物带来了突破性的综合性能。在力学性能方面,15%共聚物和30%共聚物的拉伸强度均与常规尼龙6相当,维持了工程塑料的核心优势。然而,断裂伸长率的变化令人惊叹——15%共聚物达到346%,是常规尼龙6的2.88倍;30%共聚物更是飙升至523%(图4d、e)。这种超高的柔韧性使得30%共聚物样品可以弯曲超过90度而不发生断裂,这是刚性常规尼龙6完全无法实现的变形。在光学性能方面,随着结晶度降低,共聚物的透光率从常规尼龙6的71.5%大幅提升至30%共聚物的88.9%,同时密度从1.152 g·cm⁻³降至0.908 g·cm⁻³,低于水的密度,展现出轻量化优势。照片直观地展示了随着丙基含量增加,材料从乳白不透明逐渐变为高度透明的变化过程(图4f)。研究团队将15%共聚物与常规尼龙6以及高性能商业透明尼龙12进行了雷达图对比分析。结果表明,丙基功能化共聚物成功整合了两种材料的优势:既达到了与商业透明尼龙12相当的优异透明性和低密度,又保持了常规尼龙6的高拉伸强度,同时断裂伸长率和杨氏模量均显著优于商业对照材料(图4g)。
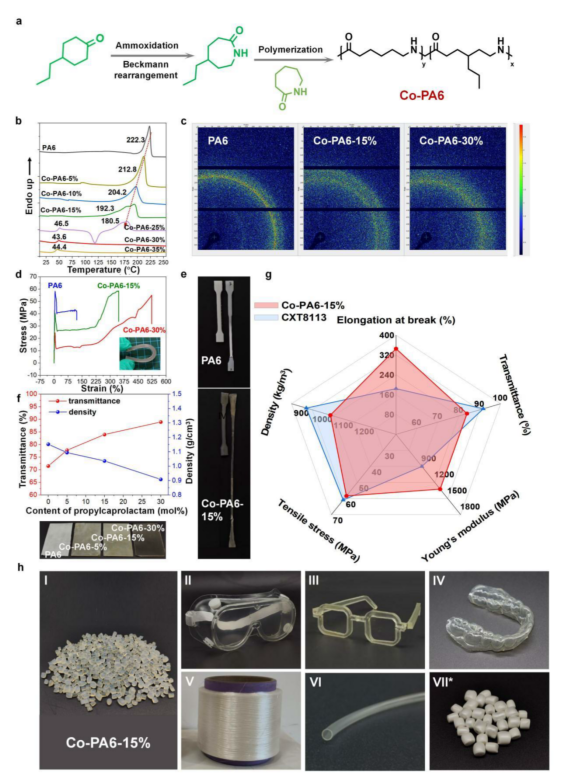
图4 | 丙基官能化共聚酰胺(Co-PA6)的合成与综合性能。 a. 4-丙基己内酰胺单体的合成路线及其与己内酰胺共聚生成Co-PA6;b. 以不同摩尔比(5-35 mol%)的4-丙基己内酰胺单体合成的PA6和Co-PA6共聚物的DSC升温和降温曲线;c. PA6和Co-PA6共聚物(Co-PA6-15%和Co-PA6-30%)的2D WAXD图案;d. PA6和Co-PA6共聚物(Co-PA6-15%和Co-PA6-30%)的拉伸应力-应变曲线;e. PA6和Co-PA6-15%的断裂伸长率(%)对比;f. 4-丙基己内酰胺单体含量对Co-PA6共聚物密度和光学透光率的影响,下方为相应照片展示透明度变化;g. Co-PA6-15%与PA6和商业化透明CXT8113关键性能对比的雷达图;h. Co-PA6-15%共聚物的产品照片:I,切片;II,抗冲击护目镜;III,透明可漂浮眼镜框架;IV,聚合物牙科隐形矫治器;V,单丝;VI,透明管材,以及Co-PA6-30%共聚物的发泡珠粒照片(VII)。
基于丙基功能化共聚物优异的综合性能,研究团队成功制备了多种高附加值产品。照片展示了15%共聚物加工的多种制品:高粘度切片、抗冲击护目镜、可漂浮于水面的低密度眼镜框架、透明牙齿矫治器、高弹性纤维以及透明管材(图4h-I至VI)。此外,利用30%功能单体含量的共聚物独特的弹性特性,研究团队通过超临界发泡获得了高回弹泡沫珠粒,适用于高性能运动器材(图4h-VII)。这些成果充分证明,木质素来源的丙基功能化己内酰胺是调控聚酰胺性能的宝贵结构单元。总结而言,本研究建立了一个精准的无氢光热催化平台用于木质素的选择性高值化转化,更重要的是,证明了烷基功能化能够赋予尼龙6前所未有的卓越性能——高透明性、高弹性、超高延伸率和低密度。该策略成功保留了木质素天然的烷基侧链并将其整合入聚合物主链,为设计高性能生物基塑料开辟了全新路径,也为实现碳中和背景下的可持续高分子材料产业提供了令人信服的范例。
专注期刊投稿、发表十年,任何投稿、写作难题欢迎咨询!

PAPER INFORMATION

快速预审、投刊前指导、专业学术评审,对文章进行评价

校对编辑、深度润色,让稿 件符合学术规范,格式体例等标准
.png)
适用于语句和结构尚需完善和调整的中文文章,确保稿件达到要求
.png)
数据库包括: 期刊、文书籍、会议、预印章、书、百科全书和摘要等
让作者在期刊选择时避免走弯路,缩短稿件被接收的周期
根据目标期刊格式要求对作者文章进行全面的格式修改和调整
帮助作者将稿件提交至目标期刊投稿系统,降低退稿或拒稿率
按照您提供的稿件内容,指导完成投稿附信(cover letter)







北京总部:北京市海淀区碧桐园 3 号楼 2 层 211 广州办事处:广州市黄埔区科学城国际企业孵化器 E栋306 联系人:客服 / 18163670350
Copyright © 2022-2024 北京特诺科技有限公司 版权所有 备案/许可证编号为: 京 ICP 备 2023007944 号
