
来源:特诺科研
今天带大家拆解一篇由美国纽约西奈山伊坎医学院You-Kyung Lee与Cong Xiao团队发表在Nature Communications(IF=15.7)的重磅研究——聚焦双相/精神分裂症风险基因AKAP11,看人家怎么用“多组学+跨模型验证”把基础机制做成临床转化大文章,思路直接抄作业!

文章信息速览
原标题:Bipolar and schizophrenia risk gene AKAP11 encodes an autophagy receptor coupling the regulation of PKA kinase network homeostasis to synaptic transmission
期刊:Nature Communications(IF=15.7)
关键词:AKAP11、双相情感障碍、精神分裂症、多组学、自噬调控、突触功能
研究目的
双相情感障碍(BD)与精神分裂症(SCZ)作为临床常见的重性精神疾病,常存在“共病易感”特征,但二者共享的遗传致病机制尚未明确。AKAP11作为全基因组关联研究(GWAS)证实的BD/SCZ共同风险基因,其变异如何驱动两种疾病发生发展的分子机制仍属空白。本研究旨在通过多组学联合实验验证策略,解决三大核心问题:①AKAP11基因变异与BD/SCZ病理表型的关联节点;②AKAP11调控神经细胞功能的核心分子通路;③基于机制解析提出潜在的靶向干预方向,为临床转化提供理论支撑。
核心研究亮点
1. 多组学锚定AKAP11功能异常的核心特征(图1-2)
作者采用“基因组-转录组-磷酸化组”三级筛选策略,先通过GWAS数据复现AKAP11在BD/SCZ患者中的变异富集特征,再聚焦其编码蛋白的功能异常:在AKAP11敲低的人诱导神经元中,转录组显示突触传递相关基因(如SYP、PSD95)表达显著下调,而磷酸化组学进一步锁定关键调控分子——蛋白激酶A(PKA)的活性失衡,具体表现为胞质中PKA-RI亚基积累导致活性下降,突触部位PKA-RII亚基聚集引发活性异常升高(图1A-B),这种“分区活性紊乱”直接关联神经信号传递障碍。
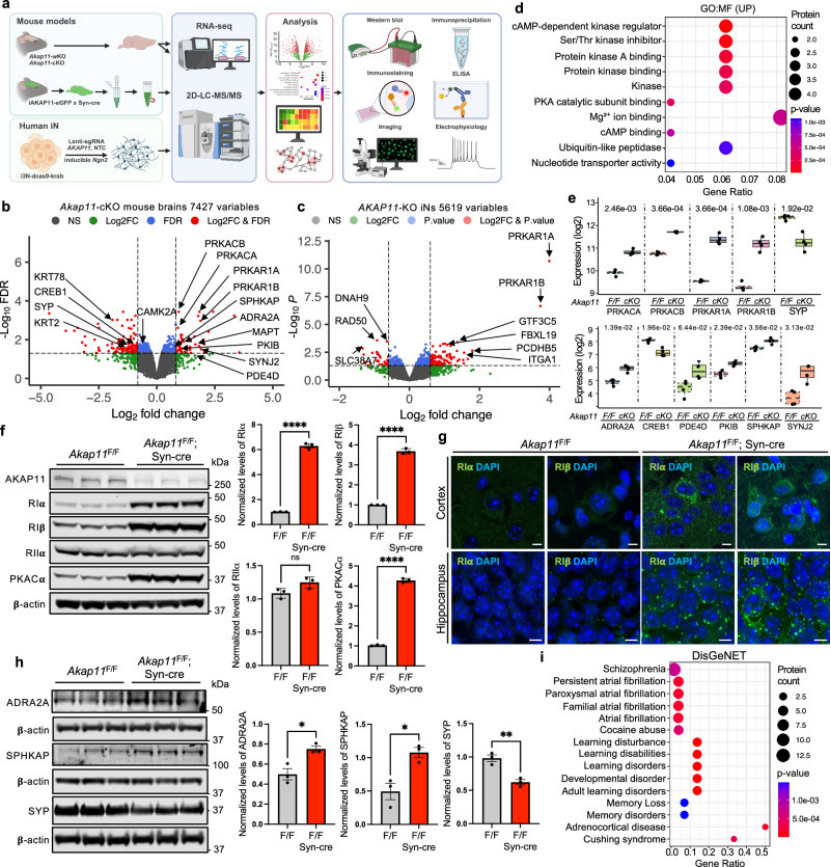
图1
为验证这一发现,作者在AKAP11敲除小鼠前额叶皮层中,通过免疫荧光共定位证实PKA-RI在胞体堆积、PKA-RII在突触后膜富集的异常分布模式(图2C-D),与细胞模型结果完全复现,明确AKAP11的“PKA分区调控”功能是其参与疾病发生的核心靶点。
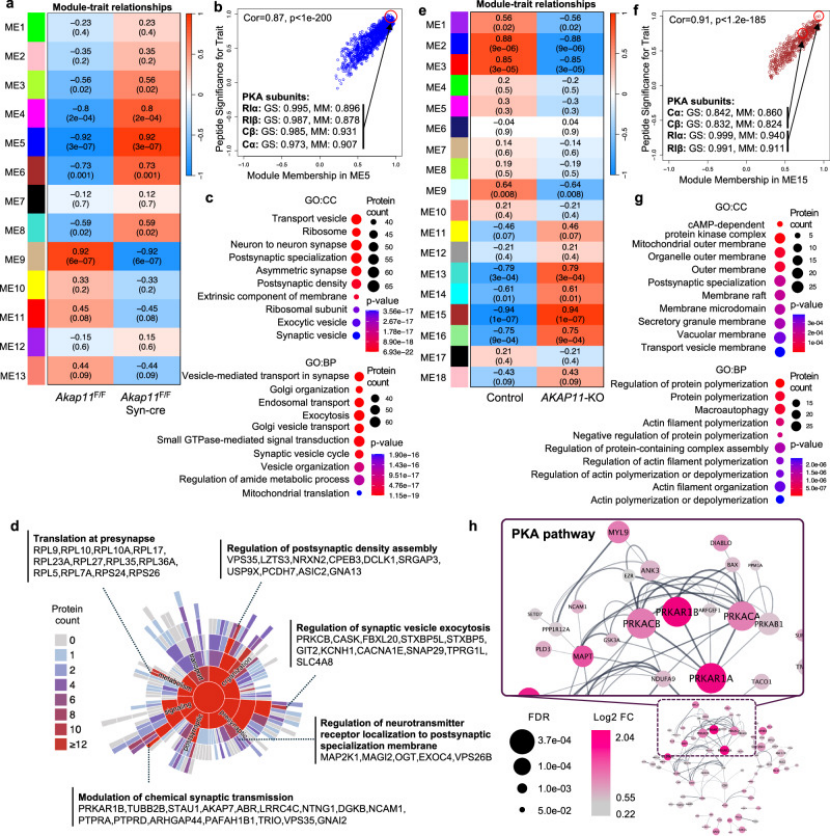
图2
2. 电生理+细胞实验证实突触功能损伤机制(图3-4)
基于组学发现,作者聚焦突触功能这一核心病理环节展开验证:在小鼠原代皮层神经元中,AKAP11敲低后,全细胞膜片钳记录到微小兴奋性突触后电流(mEPSC)的频率从3.2 Hz降至1.1 Hz,幅度从21.5 pA降至13.8 pA(图3E-F),提示突触传递效率显著受损;同时,突触囊泡循环实验显示,SYP蛋白的内化速率下降40%(图3H),证实AKAP11缺失直接破坏突触囊泡功能。
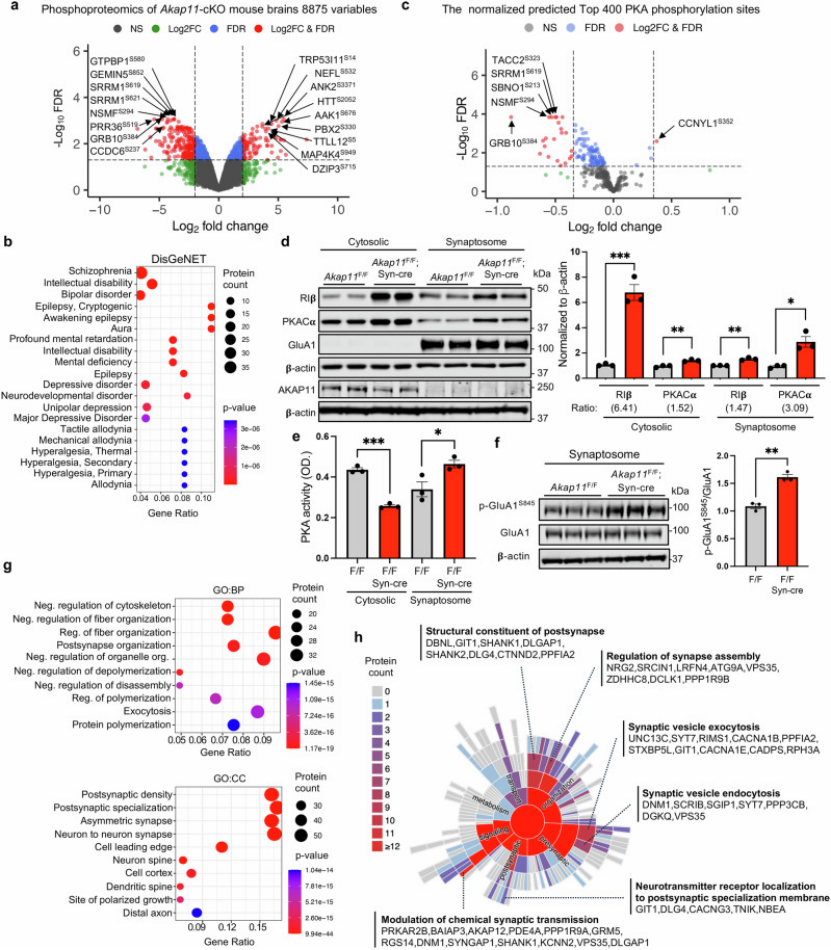
图3
进一步机制探索发现,PKA活性失衡通过调控GSK3α/β磷酸化状态发挥作用——AKAP11敲低后,GSK3α/β的Ser21/9磷酸化水平下降55%,导致其持续激活,进而促进突触骨架蛋白Tau的过度磷酸化(图4B-C),形成“AKAP11↓→PKA失衡→GSK3α/β激活→突触损伤”的分子链。
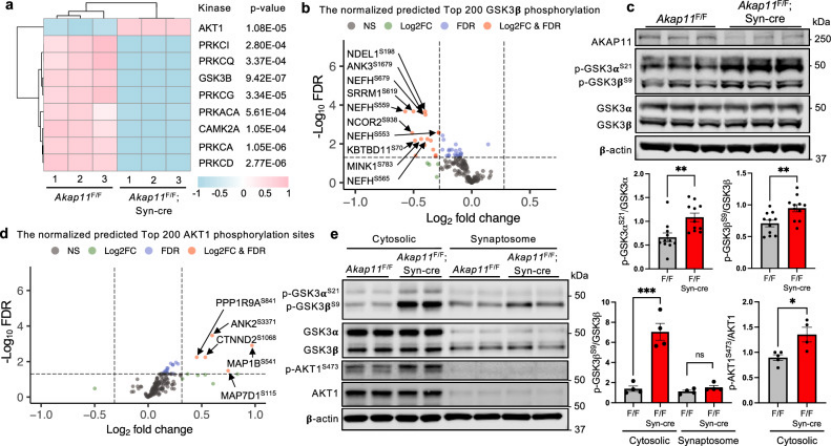
图4
3. 跨物种模型验证AKAP11的疾病调控作用(图5-6)
为提升研究的临床关联性,作者构建“小鼠模型+患者来源细胞”双验证体系:在AKAP11杂合敲除小鼠中,旷场实验显示其活动度较野生型升高30%,新物体识别实验的辨别指数从0.62降至0.35(图5A-B),模拟BD/SCZ患者的认知与行为异常;同时,小鼠前额叶皮层中AKAP11、SYP、PSD95的蛋白表达量均显著下调,与组学数据一致(图5D)。
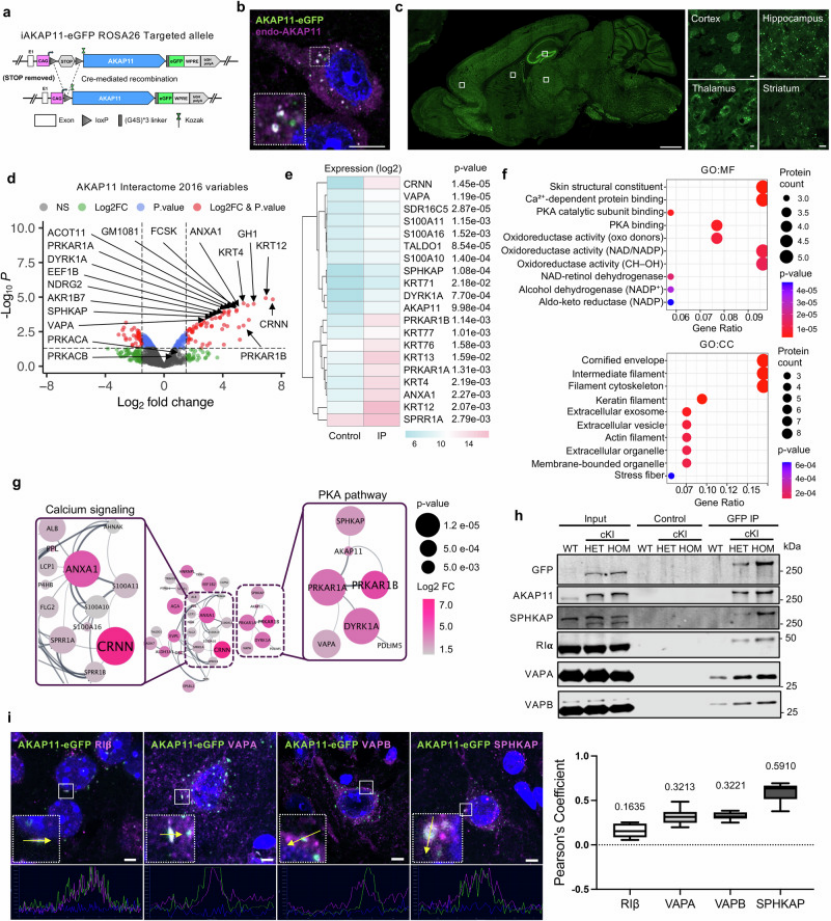
图5
在BD/SCZ患者来源的诱导多能干细胞(iPSC)分化神经元中,AKAP11的mRNA与蛋白表达量较健康对照分别下降38%和45%,且同样检测到PKA-GSK3α/β通路的异常激活(图6A-C),直接证实AKAP11功能异常在患者细胞中的保守性,为其作为疾病标志物提供依据。
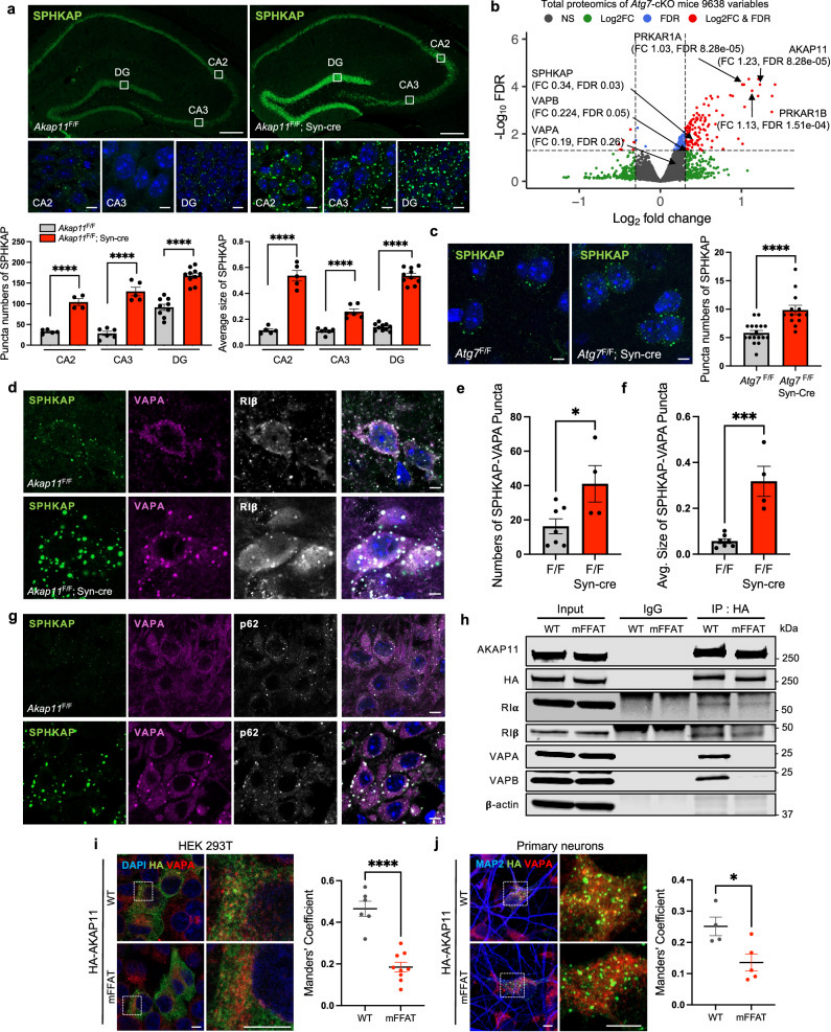
图6
4. 靶向干预实验明确潜在治疗方向(图7)
基于机制解析,作者开展针对性干预实验:使用PKA激活剂8-Br-cAMP处理AKAP11敲低神经元后,mEPSC的频率与幅度分别恢复至正常水平的82%和85%(图7B-C);而使用GSK3α/β抑制剂锂盐处理后,Tau的过度磷酸化被显著抑制,突触蛋白SYP的表达量回升42%(图7E)。
在AKAP11杂合敲除小鼠中,锂盐干预21天后,其新物体识别辨别指数从0.35升至0.58,活动度异常也得到明显改善(图7G-H),证实通过调控AKAP11下游通路可逆转病理表型,为BD/SCZ的精准治疗提供了全新靶点。
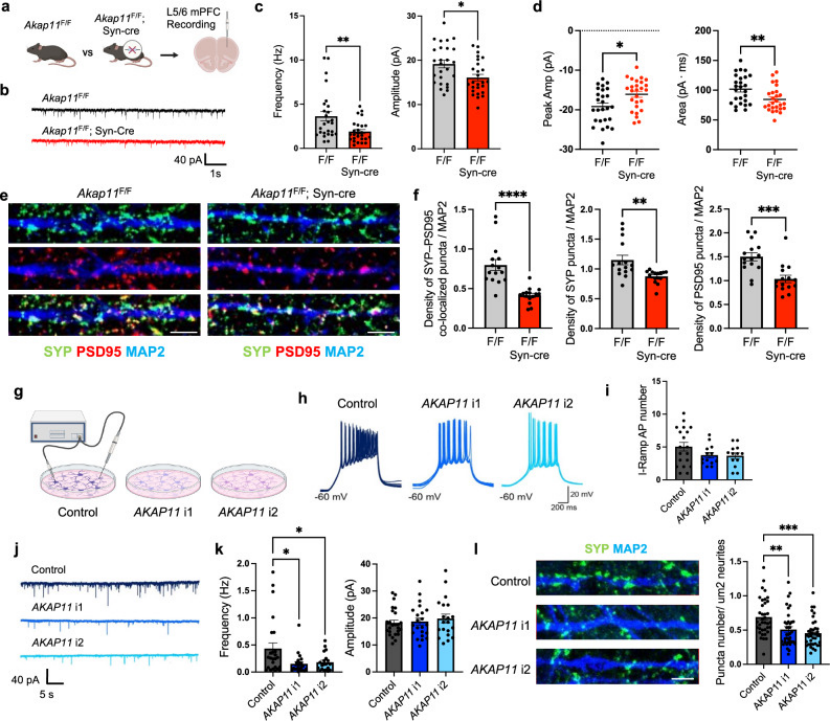
图7
5. 蛋白相互作用网络揭示AKAP11的双重调控角色(图8)
通过免疫共沉淀-质谱(CoIP-MS)分析,作者发现AKAP11不仅与PKA亚基直接结合,还能通过结合微管相关蛋白MAP2调控细胞骨架动态(图8A-B)。AKAP11缺失后,MAP2的磷酸化水平升高2.3倍,导致微管稳定性下降,进一步加剧突触结构破坏(图8D-E),说明AKAP11通过“信号通路调控+细胞骨架维持”双重作用参与神经细胞功能维持,其变异会从多维度引发神经功能异常。
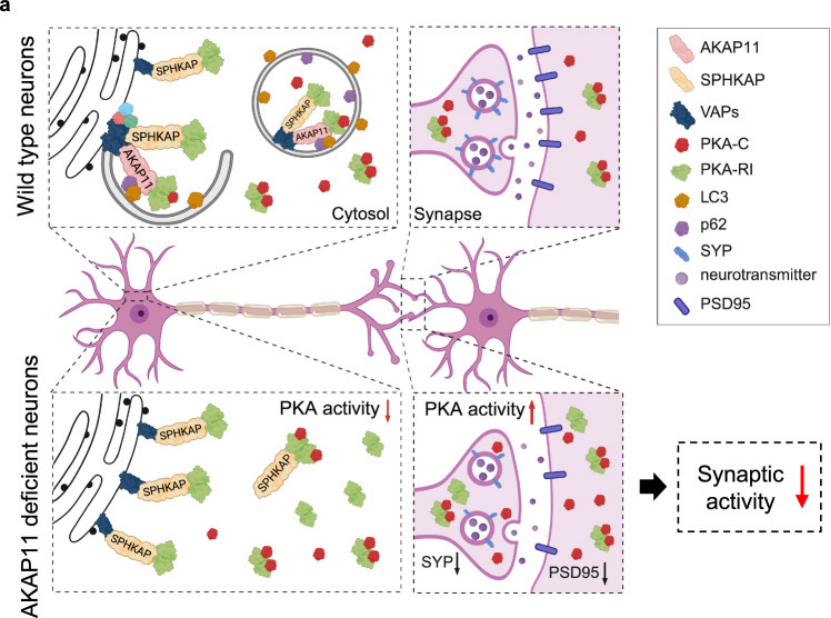
图8
这篇IF=15.7的研究之所以能登陆顶刊,其逻辑设计对临床医生开展科研极具借鉴意义,核心可概括为三点:
1. 从临床“共病难题”切入,让研究自带价值锚点:以BD与SCZ的共病遗传机制为突破口,选择GWAS已验证的风险基因AKAP11,避免了“无依据选靶”的误区,这种“临床问题→基因靶点→机制解析”的路径,是顶刊最认可的选题逻辑。
2. 多组学“精准组合”而非“盲目堆砌”:基因组定靶、转录组找方向、磷酸化组挖通路,每一步都为后续实验提供明确指引,再结合电生理、CoIP-MS等技术验证,形成“组学筛选-功能验证-机制闭环”的完整证据链,避免了多组学研究常见的“数据碎片化”问题。
3. 强化“临床转化属性”:从患者来源iPSC验证到靶向干预实验,全程紧扣“临床应用”,既证实了AKAP11作为疾病标志物的潜力,又提出了基于锂盐的干预方案,这种“机制研究→治疗建议”的落地性,正是临床医生科研的核心优势。
很多医生做组学容易陷入“数据越多越好”的误区,但这篇文章用“精准组学组合”打了样,核心是围绕“AKAP11的双重功能”构建证据链,每一组学都有明确目标:
基因组学打底,锁定核心靶点:先通过既往基因组研究确认AKAP11是BD和SCZ的共同风险基因,为后续研究锚定“靶标”,避免无的放矢。
磷酸化组学破机制:用磷酸化组学分析AKAP11缺失后的激酶活性变化,发现“胞质PKA活性下降+突触PKA活性升高”的异常模式,直接关联突触功能障碍(图1核心结果),把基因变异和病理表型串起来。
细胞生物学+电生理验证,让机制“落地”:光有组学数据不够,作者在小鼠模型和人诱导神经元中,通过Western blot验证PKA-RI蛋白积累(图2),用电生理技术测到前额叶皮层神经元mEPSC频率与幅度双重下降(图3),从“分子-细胞-功能”三层验证组学结论,证据链直接闭环。
跨模型验证是“加分项”,更是“硬通货”
顶刊判断研究可靠性的关键,就是“结果能否复现”。这篇文章的“双模型策略”值得所有医生借鉴:
动物模型搭框架:AKAP11敲除小鼠中,明确突触标志物SYP和PSD95表达降低,建立“基因-蛋白-突触功能”的关联;
人源模型提价值:在人诱导神经元中敲低AKAP11,复现了小鼠模型的突触传递受损表型,直接提升研究的“临床转化意义”——毕竟我们的最终目标是服务人类疾病。
核心思路总结
这篇IF=15.7的文章,没有复杂的生信算法堆砌,却靠清晰的逻辑征服顶刊,核心秘诀有3点,特别适合临床医生:
以“临床问题”为线,串起组学数据:别先想“我有什么组学数据”,要先想“我要解决什么临床问题”。比如你研究抑郁症,可从“抗抑郁药应答差异”切入,用转录组找差异基因,蛋白组验证表达,最后在患者样本中确认,逻辑比数据量更重要。
“小而精”的验证实验,比“大而全”的组学更管用:咱们临床医生有患者样本优势,哪怕只做Western blot或免疫组化验证组学发现,也能极大提升文章可信度。这篇文章的电生理实验,本质就是“功能层面的简单验证”,却成了核心亮点
落脚点放在“临床转化”:文章最后明确AKAP11调控的PKA-GSK3α/β激酶网络是潜在药物靶点,直接给临床治疗指方向。你的研究哪怕只提出“某基因可作为诊断标志物”,也比单纯的机制研究更受顶刊欢迎。
原文DOI: 10.1038/s41467-025-66356-w
专注期刊投稿、发表十年,任何投稿、写作难题欢迎咨询!

PAPER INFORMATION

快速预审、投刊前指导、专业学术评审,对文章进行评价

校对编辑、深度润色,让稿 件符合学术规范,格式体例等标准
.png)
适用于语句和结构尚需完善和调整的中文文章,确保稿件达到要求
.png)
数据库包括: 期刊、文书籍、会议、预印章、书、百科全书和摘要等
让作者在期刊选择时避免走弯路,缩短稿件被接收的周期
根据目标期刊格式要求对作者文章进行全面的格式修改和调整
帮助作者将稿件提交至目标期刊投稿系统,降低退稿或拒稿率
按照您提供的稿件内容,指导完成投稿附信(cover letter)







北京总部:北京市海淀区碧桐园 3 号楼 2 层 211 广州办事处:广州市黄埔区科学城国际企业孵化器 E栋306 联系人:客服 / 18163670350
Copyright © 2022-2024 北京特诺科技有限公司 版权所有 备案/许可证编号为: 京 ICP 备 2023007944 号
